Parameters¶
The NextITS pipeline is made up of multiple connected processes.
Each of these processes is amenable to configuration,
empowering users to tailor not just the process parameters
but also the resources allocated for each one (e.g., the number of CPUs).
When setting the pipeline parameter values, users have two choices:
they can specify them directly from the command line using double dash arguments (for example, --its_region "full")
or opt for a simple parameters file. Utilizing a parameters file is especially advantageous
when dealing with numerous settings, as it offers a cleaner command line interface.
In addition to this, individual processes within the pipeline can be fine-tuned using a dedicated config file.
NextITS boasts a vast array of configurable parameters. For ease of navigation and understanding, these parameters have been grouped into several categories, detailed in the sections that follow.
Step-1¶
Required parameters¶
| Parameter | Description |
|---|---|
--input |
Path to the input file containing single-end sequences (in FASTQ format) or the directory with pre-demultiplexed files |
--barcodes |
Path to the file with barcodes (in FASTA format) used for demultiplexing the input data |
--outdir |
Path to the directory where the analysis results will be saved. |
The pipeline can handle both multiplexed and demultiplexed data inputs:
For multiplexed data:
- Use the --input parameter to specify a single FASTQ file (e.g., Run01.fastq.gz);
- You must also provide the --barcodes parameter, which should reference a FASTA file. The sequence names in this file should correspond to the sample names.
For pre-demultiplexed data (prepared using external tools):
- The --input parameter should direct to a directory containing individual FASTQ files for each sample;
- There's no need to provide a barcodes file in this case;
- Demultiplexed parameter should be enabled (--demultiplexed true).
Demultiplexing¶
| Parameter | Description | Default Value |
|---|---|---|
--demultiplexed |
Whether input is multiplexed (false, single FASTQ file) or pre-demultiplexed (true, multiple FASTQ files) 1 |
false |
--lima_barcodetype |
Barcoding scheme type (single, dual, dual_symmetric, dual_asymmetric) 2 |
dual_symmetric |
--lima_minscore |
Barcode score for demultiplexing 3 | 93 |
--lima_W |
Window size for barcode lookup | 70 |
--lima_minlen |
Minimum sequence length after clipping barcodes | 40 |
--lima_minendscore |
Score threshold for asymmetric barcoding schemes with different barcodes in a pair | 50 |
--lima_minrefspan |
Minimum read span relative to the barcode length | 0.75 |
--lima_minscoringregions |
Number of required barcodes for dual barcodes (2 = requires both barcodes) | 2 |
--lima_windowsize |
Window size in base pairs | 70 |
--lima_remove_unknown |
Remove unknown barcode combinations in dual-barcoding modes | false |
1:
By default, NextITS assumes you're providing multiplexed data,
which means a single FASTQ file accompanied by a FASTA file containing barcodes.
If your data is already separated into individual samples
(meaning you have multiple FASTQ files, potentially more than one for each sample),
use the --demultiplexed true option.
2:
Barcodes, which can also be referred to as adaptors, tags, indices, molecular identifiers (MIDs), come in various library designs.
At present, NextITS supports 4 barcoding schemes: dual-barcoding (symmetric, assymetric, or mixture of combinatorial tags) and single-barcoding.
For the dual-barcoding scheme, the amplicon should have the barcode on both ends of the amplicon.
An asymmetric design, where each side of the amplicon has a different barcode pair, is also supported.
For a comprehensive understanding of barcode designs, please visit https://lima.how/barcode-design.html.
3:
For every barcode, LIMA evaluates a score corresponding to its region.
This score signifies the alignment accuracy between the chosen barcode and sequencing read,
and it's determined using the Smith-Waterman algorithm.
For the HiFi data, adjust the score based on your tolerance for contamination:
a setting of --min-score 80 should ensure over 99.99% precision.
If you're working with shorter barcodes, such as 10 base pairs, consider reducing the minimum score for optimal results.
For a more comprehensive information, check out https://lima.how/faq/barcode-score.html.
Quality filtering¶
| Parameter | Description | Default Value |
|---|---|---|
--qc_maxee |
Maximum number of expected errors 1 | - |
--qc_maxeerate |
Maximum number of expected errors per base | 0.01 |
--qc_maxhomopolymerlen |
Threshold for a homopolymer region length in a sequence 2 | 25 |
--qc_maxn |
Discard sequences with more than the specified number of ambiguous nucleotides (N's) | 4 |
1:
As part of the sequence quality control, it is possible to evaluates sequences based on the
maximum number of expected errors (MaxEE) predicted by Phred scores
(Edgar & Flyvbjerg 2015 DOI:10.1093/bioinformatics/btv401)
and the maximum number of expected errors per base.
2:
The ITS region of many fungal and other eukaryotic taxa could commonly harbour homopolymers exceeding 10 bases
(Tedersoo et al. 2022 DOI:10.1111/mec.16460).
Removal of multiprimer-artifacts and reorienting of reads¶
| Parameter | Description | Default Value |
|---|---|---|
--primer_forward |
Sequence of the forward primer 1 | TACACACCGCCCGTCG |
--primer_reverse |
Sequence of the reverse primer 1 | CCTSCSCTTANTDATATGC |
--primer_mismatches |
Allowed number of mismatches for primers | 2 |
--primer_foverlap |
Minimum overlap for the forward primer 2 | F primer length - 2 |
--primer_roverlap |
Minimum overlap for the reverse primer 2 | R primer length - 2 |
Default Primers:
By default, NextITS is configured for the data obtained with universal eukaryote primers:
- Forward Primer: ITS9MUNngs with sequence TACACACCGCCCGTCG
- Reverse Primer: ITS4ngsUni with sequence CCTSCSCTTANTDATATGC
Refer to Tedersoo & Lindahl, 2016 DOI:10.1111/1758-2229.12438 for more details on these primers.
Alternative Forward Primer:
For specific applications, we also recommend the ITS1catta forward primer (sequence: ACCWGCGGARGGATCATTA).
It's designed to target and exclude plant sequences while avoiding interference from the SSU 3′-end intron.
For additional information on this primer, consult Tedersoo & Anslan, 2019 DOI:10.1111/1758-2229.12776.
ITS extraction¶
| Parameter | Description | Default Value |
|---|---|---|
--its_region |
ITS part selector 2 | "full" |
--ITSx_tax |
Taxonomy profile for ITSx 1 | "all" |
--ITSx_evalue |
E-value cutoff threshold for ITSx | 1e-1 |
--ITSx_partial |
Keep partial ITS sequences (specify a minimum length cutoff), default = off (0) | 0 |
When performing ITS metabarcoding, it's essential to trim the flanking 18S and 28S rRNA genes.
This is crucial because:
- These conserved regions don't offer species-level differentiation.
- Random errors in these areas can disrupt sequence clustering.
- Chimeric breakpoints, which are common in these regions, are hard to detect in short fragments ranging from 10 to 70 bases.
(For details, see Lindahl et al. 2013 DOI:10.1111/nph.12243 and
Tedersoo et al. 2022 DOI:10.1111/mec.16460).
To address this, NextITS employs the ITSx program (Bengtsson-Palme et al., 2013 DOI:10.1111/2041-210X.12073).
By using the --its_region parameter, users can select the region for subsequent analyses.
Supported options include:
- full: Represents the full-length ITS
- ITS1_5.8S_ITS2: A near-full-length ITS that assembles the sequence from the ITS1, 5.8S, and ITS2 segments extracted by ITSx.
This option is particularly handy when ITSx struggles to detect the end of the SSU.
(for instance, when using the ITS1catta primer, which is located at the extreme end of the SSU and is not detected by ITSx).
- ITS1: Represents the ITS1 region
- ITS2: Represents the ITS2 region
- SSU: Focuses on the small subunit of rRNA (18S)
- LSU: Focuses on the large subunit of rRNA (28S)
- none: This option only trims primers without extracting the ITS
At the ITS extraction step of the pipeline, you can define a specific taxonomic group of interest.
ITSx employs hidden Markov models (HMMs) tailored for 20 eukaryotic taxonomic groups.
These models detect patterns in rRNA by recognizing variations commonly observed in multiple sequence alignments.
To select the taxon of interest, use the --ITSx_tax parameter.
You can input either single-character codes (e.g., --ITSx_tax f) or full names (e.g., --ITSx_tax fungi).
For multiple selections, provide a comma-separated string, such as --ITSx_tax amoebozoa,rhizaria.
For mixed-taxon datasets, there's an all-encompassing search option: --ITSx_tax all (which is enabled by default).
A table detailing all supported organism groups is provided below:
| Code | Full name | Alternative name |
|---|---|---|
. |
All | all |
A |
Alveolata | alveolates |
B |
Bryophyta | mosses |
C |
Bacillariophyta | diatoms |
D |
Amoebozoa | |
E |
Euglenozoa | |
F |
Fungi | |
G |
Chlorophyta | green-algae |
H |
Rhodophyta | red-algae |
I |
Phaeophyceae | brown-algae |
L |
Marchantiophyta | liverworts |
M |
Metazoa | animals |
O |
Oomycota | oomycetes |
P |
Haptophyceae | prymnesiophytes |
Q |
Raphidophyceae | raphidophytes |
R |
Rhizaria | |
S |
Synurophyceae | synurids |
T |
Tracheophyta | higher-plants |
U |
Eustigmatophyceae | eustigmatophytes |
X |
Apusozoa | |
Y |
Parabasalia | parabasalids |
Chimera identification¶
| Parameter | Description | Default Value |
|---|---|---|
--chimera_db |
Database for reference-based chimera removal (UDB) | - |
--chimera_rescueoccurrence |
Min occurrence of chimeric sequences required to rescue them | 2 |
--chimeranov_abskew |
abskew parameter for de novo chimera identification |
2.0 |
--chimeranov_dn |
dn parameter for de novo chimera identification |
1.4 |
--chimeranov_mindiffs |
mindiffs parameter for de novo chimera identification |
3 |
--chimeranov_mindiv |
mindiv parameter for de novo chimera identification |
0.8 |
--chimeranov_minh |
minh parameter for de novo chimera identification |
0.28 |
--chimeranov_xn |
xn parameter for de novo chimera identification |
8.0 |
NextITS employs a two-pronged strategy to detect chimeras:
-
De novo Detection:
This algorithm identifies chimeras without relying on reference data. To mitigate the risk of false-positive detections, sequences flagged as chimeras by the de novo method are not discarded instantly. Instead, they're assigned a chimeric score. Filtering based on this score can be conducted in subsequent analysis stages (refer to Step-2 for more details). -
Reference-based Detection:
This method leverages a reference database in the UDB format.
An example of such a database is accessible at: https://owncloud.ut.ee/owncloud/s/iaQ3i862pjwYgdy.
Furthermore, NextITS offers an option to "rescue" sequences mistakenly labeled as chimeras.
The pipeline checks if these sequences appear in other samples where they are not identified as chimeras.
If a sequence is consistently observed, it's more likely to be a genuine biological sequence.
You can control this functionality using the --chimera_rescueoccurrence parameter.
Homopolymer correction¶
| Parameter | Description | Default Value |
|---|---|---|
--hp |
Enable or disable homopolymer correction | true |
--hp_similarity |
Allowed sequence similarity for homopolymer correction | 0.999 |
--hp_iddef |
Sequence similarity definition for homopolymer correction | 2 |
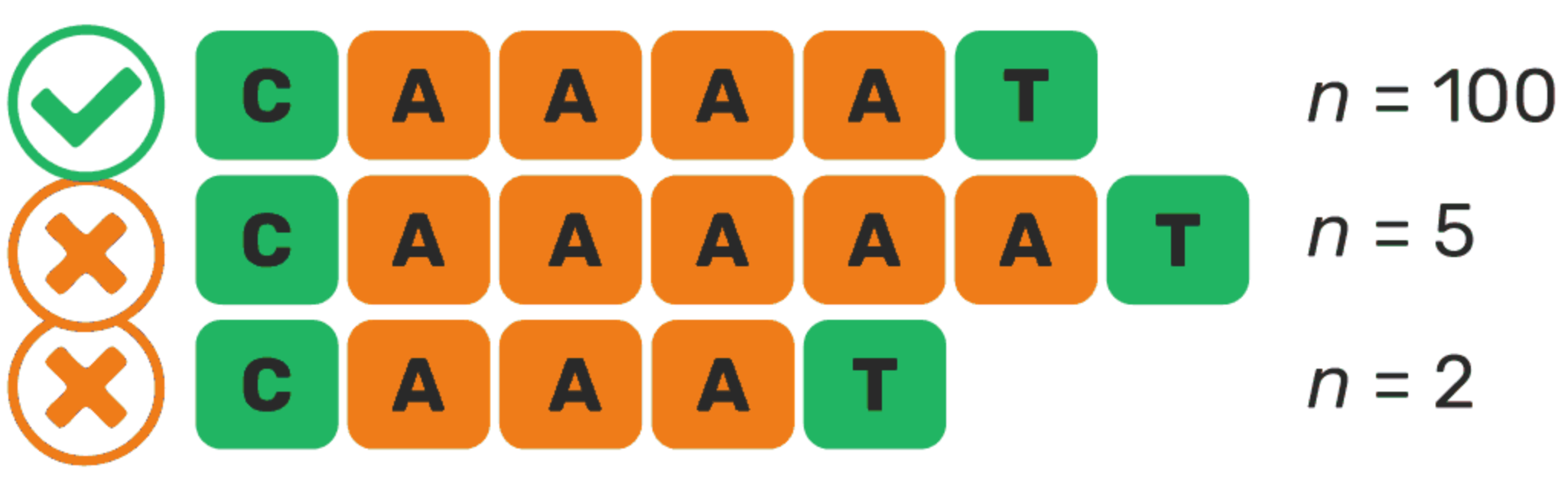
Although the PacBio HiFi reads are highly accurate, but they sometimes show errors in homopolymeric sites (e.g., AAAAAAA stretches).
These sites are also naturally prevalent in fungal ITS.
Such errors can lead to an inflated number of OTUs, slowing down clustering and reducing its efficiency.
To counteract this, NextITS has a strategy to correct these homopolymer errors.
For each sample, the dominant variant of the homopolymer sequence is retained and used.
Tag-jump Removal Parameters¶
| Parameter | Description | Default Value |
|---|---|---|
--tj_f |
UNCROSS parameter f for tag-jump filtering |
0.01 |
--tj_p |
Parameter p for tag-jump filtering |
1 |
--otu_id |
Sequence similarity for OTU clustering | 0.98 |
--otu_iddef |
Sequence similarity definition for tag-jump removal step | 2 |
Tag-jumps, sometimes referred to as index-switches or index cross-talk, are significant concerns in high-throughput sequencing (HTS) data
(Tedersoo et al. 2022 DOI:10.1111/mec.16460).
They can cause technical cross-contamination between samples, potentially distorting estimates of microbial community composition.
While careful sample indexing can mitigate this problem, a small percentage (approximately 0.01–0.1%) of these errors might persist in the data.
NextITS provides a solution to evaluate index-switches using the
UNCROSS2 algorithm
(Edgar 2018 DOI:10.1101/400762),
which assigns an ad hoc score to measure the likelihood of tag-jump events.
Step-2¶
Required parameters¶
| Parameter | Description |
|---|---|
--data_path |
Input directory containing the results of Step-1 |
--outdir |
Path to the directory where the analysis results will be saved. |
Denoising parameters (optional)¶
| Parameter | Description | Default Value |
|---|---|---|
--unoise |
Perform denoising with UNOISE algorithm | false |
--unoise_alpha |
Alpha parameter of UNOISE 1 |
2.0 |
--unoise_minsize |
Minimum sequence abundance 2 | 8 |
The UNOISE algorithm (Edgar, 2016, DOI:10.1101/081257)
focuses on error-correction (or denoising) of amplicon reads.
Essentially, UNOISE operates on the principle that if a sequence with low abundance
closely resembles another sequence with high abundance, the former is probably an error.
This helps differentiate between true biological variation and sequencing errors.
1:
The --unoise_alpha option sets the alpha parameter, as described in the UNOISE2 paper.
Essentially, this parameter establishes the dissimilarity threshold between frequent and infrequent reads.
2:
The --unoise_minsize option (which corresponds to the -minsize parameter in USEARCH),
determines the minimum abundance a sequence must have across all samples.
Any sequences with abundances below this threshold will be discarded, as sequences with very low abundances tend to be noisy.
It's important to note that UNOISE was initially designed and optimized for Illumina data.
Because of indel errors stemming from inaccuracies in homopolymeric regions,
UNOISE might not work well with data that hasn't undergone homopolymer correction
(more details at the USEARCH web-site).
Therefore, when using UNOISE, ensure you activate both options by setting --unoise true and --hp true together.
Sequence clustering¶
| Parameter | Description | Default Value |
|---|---|---|
--clustering_method |
Sequence clustering method | "vsearch" |
Supported Methods:
- vsearch:
This employs greedy clustering using a fixed sequence similarity threshold with
VSEARCH
(Rognes et al., 2016, DOI:10.7717/peerj.2584);
- swarm:
Unlike "vsearch", this uses a dynamic sequence similarity threshold for clustering with
SWARM
(Mahé et al., 2021, DOI:10.1093/bioinformatics/btab493);
- unoise:
This focuses solely on denoising to create zero-radius OTUs (zOTUs)
based on the UNOISE3 algorithm
(Edgar, 2016, DOI:10.1101/081257);
- shmatching (currently under development):
This method clusters sequences based on species hypotheses (SH) as detailed in
(Kõljalg et al., 2013, DOI:10.1111/mec.12481,
Abarenkov et al., 2022, DOI:10.3897/biss.6.93856
);
VSEARCH clustering¶
| Parameter | Description | Default Value |
|---|---|---|
--otu_id |
Sequence similarity threshold | 0.98 |
--otu_iddef |
Sequence similarity definition (applied to UNOISE as well) | 2 |
--otu_qmask |
Method to mask low-complexity sequences (applied to UNOISE as well) | "dust" |
SWARM clustering¶
| Parameter | Description | Default Value |
|---|---|---|
--swarm_d |
SWARM clustering resolution (d) |
1 |
--swarm_fastidious |
Link nearby low-abundance swarms (fastidious option) |
true |
--swarm_d1boundary |
Minimal mass of large OTUs (only for fastidious with d=1) |
3 |
OTU table preparation, thresholds for singleton and de novo chimera removal¶
| Parameter | Description | Default Value |
|---|---|---|
--merge_replicates |
Pool sample replicates (e.g., re-sequenced samples) in the final OTU table | false |
--max_MEEP |
Maximum allowed number of expected errors per 100 bp | 0.5 |
--max_ChimeraScore |
Maximum allowed de novo chimera score | 0.6 |
--recover_lowqsingletons |
Logical, Recover de novo chimeras 1 | true |
--recover_denovochimeras |
Logical, Recover singletons 2 | true |
1:
Do-novo chimera recovery:
if a sequence identified as putative chimera was observed in the other samples,
where there is no evidence that it is chimeric, it will be recovered.
2:
Singleton recovery:
if a within-sequencing run singleton sequence with relatively low quality (based on MEEP)
was observed in the other samples, it will be recovered.
Post-clustering curation with LULU¶
| Parameter | Description | Default Value |
|---|---|---|
--lulu |
Run LULU | true |
--lulu_match |
Minimum similarity threshold | 95.0 |
--lulu_ratio |
Minimum abundance ratio | 1.0 |
--lulu_ratiotype |
Abundance ratio type - "min" or "avg" | "min" |
--lulu_relcooc |
Relative co-occurrence | 0.95 |
--lulu_maxhits |
Maximum number of hits (0 = unlimited) | 0 |
Please Note: The default value for --lulu_match
in NextITS is set to a 95% similarity threshold.
This differs from the original LULU paper, where the default threshold is 84%.
Miscellaneous parameters¶
| Parameter | Description | Default Value |
|---|---|---|
--gzip_compression |
Compression level for GZIP 1 | 7 |
--storagemode |
Adjusts how files are directed to the results folder 2 | "symlink" |
1:
To save disk space and potentially enhance data reading speed,
NextITS compresses data using gzip.
Adjust the balance between speed and compression ratio with the --gzip_compression parameter.
Where -1 is the fastest option (worst compression), and -9 is slowest (best compression).
2:
In the default setup, files are directed to the results folder (--output)
using symbolic links (or symlinks) which help save disk space.
Each symlink points to the actual file located in the process working directory.
This behavior can be modified using the --storagemode parameter.
Refer to the table below for a complete list of available storage mode options.
| Storage mode | Description |
|---|---|
symlink |
Creates a symbolic link to the file |
copy |
Copies the file to the target directory |
move |
Moves the file to the target directory (not recommended) |
rellink |
Creates a relative symbolic link to the file |
link |
Creates a hard link to the file |